Wastewater RNA Read Lengths |
November 10th, 2023 |
| bio, nao |
What I'm calling "read length" here should instead have been "insert length" or "post-trimming read length". When you just say "read length" that's usually understood to be the raw length output by the sequencer, which for short-read sequencing is determined only by your target cycle count.
Let's say you're collecting wastewater and running metagenomic RNA sequencing, with a focus on human-infecting viruses. For many kinds of analysis you want a combination of a low cost per base and more bases per sequencing read. The lowest cost per base these days, by a lot, comes from paired-end "short read" sequencing (also called "Next Generation Sequencing", or "Illumina sequencing" after the main vendor), where an observation looks like reading some number of bases (often 150) from each end of a nucleic acid fragment:
+------>>>-----+
| forward read |
+------>>>-----+
... gap ...
+------<<<-----+
| reverse read |
+------<<<-----+
Now, if the fragments you feed into your sequencer are short you can instead get something like:
+------>>>-----+
| forward read |
+------>>>-----+
+------<<<-----+
| reverse read |
+------<<<-----+
That is, if we're reading 150 bases from each end and our fragment is only 250 bases long, we have a negative "gap" and we'll read the 50 bases in the middle of the fragment twice.
And if the fragments are very short, shorter than how much you're reading from each ends, you'll get complete overlap (and then read through into the adapters):
+------>>>-----+ | forward read | +------>>>-----+ +------<<<-----+ | reverse read | +------<<<-----+
One shortcoming of my ascii art is it doesn't show how the effective read length changes: in the complete overlap case it can be quite short. For example, if you're doing 2x150 sequencing you're capable of learning up to 300bp with each read pair but if the fragment is only 80bp long then you'll just read the same 80bp from both directions. So overlap is not ideal from either the perspectives of minimizing cost per base or maximizing the length of each observation: a positive gap is better than any overlap, and more overlap is worse.
One important question, however, is whether this is something we can have much control over. Is RNA in wastewater just so badly degraded that there's not much you can do, or can you prepare the wastewater for sequencing in a way that minimizes additional fragmentation? The best way to answer this would be to run some head-to-head comparisons, sequencing the same wastewater with multiple candidate techniques, but what can I do just re-analyzing existing data?
As part of putting together the NAO's relative abundance report I'd already identified four studies that had conducted untargeted metagenomic RNA sequencing of wastewater and made their data public on the SRA:
Crits-Christoph et. al 2021: SF, summer 2020, 300M read pairs, 2x75.
Rothman et. al 2021: LA, fall 2020, 800M read pairs (unenriched only), 2x100 and 2x150.
Spurbeck et. al 2023: Ohio, winter 2021-2022, 1.8B read pairs, 2x150.
Yang et. al 2020: Urban Xinjiang China, 2018, 1.4B read pairs, 2x150.
After running them through the NAO's pipeline (trimming adapters, assigning species with Kraken2) I plotted the length distributions of the viral reads:
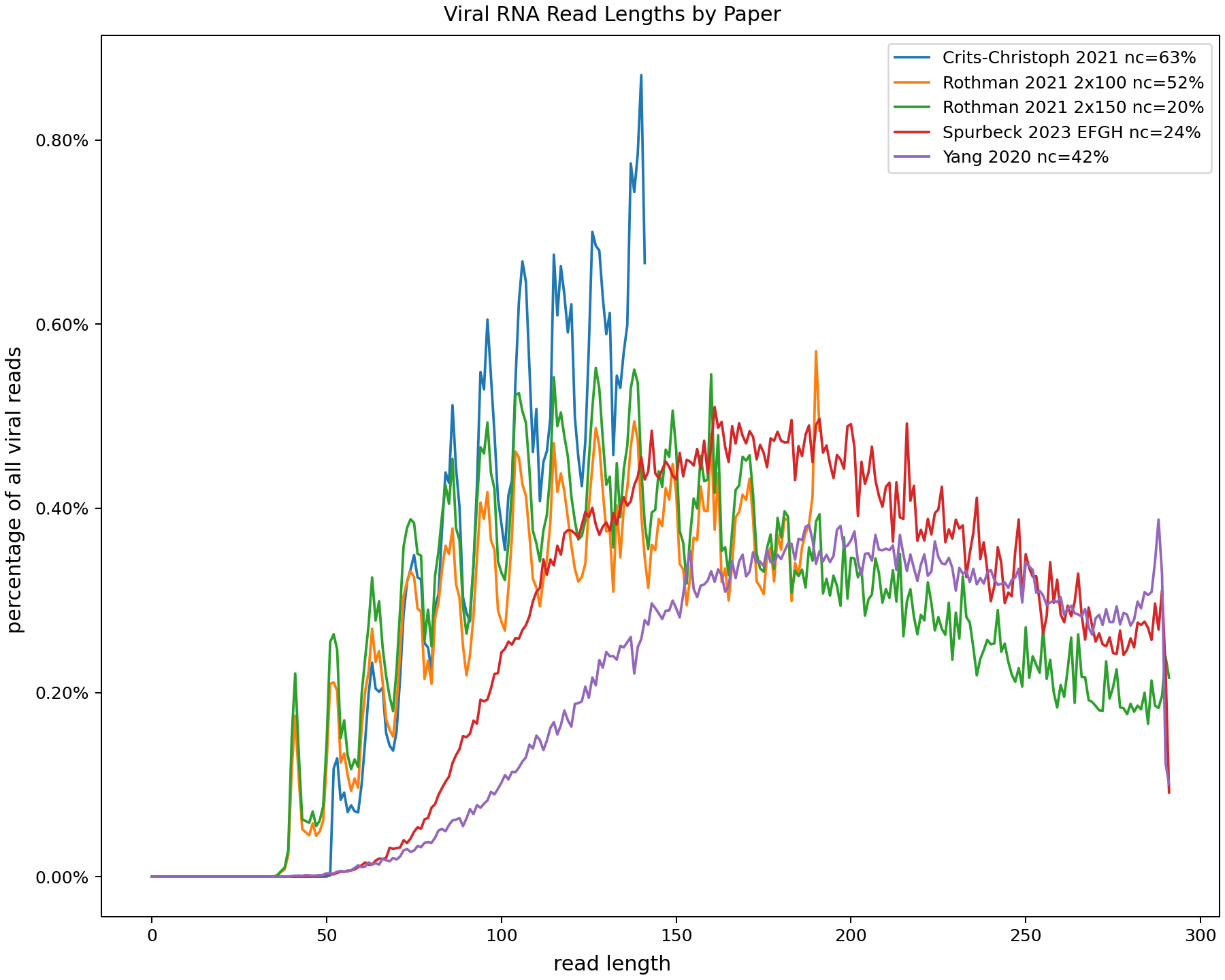
This is a little messy, both because it's noisy and because something about the process gives oscilations in the Crits-Christoph and Rothman data. [1] Here's a smoothed version:
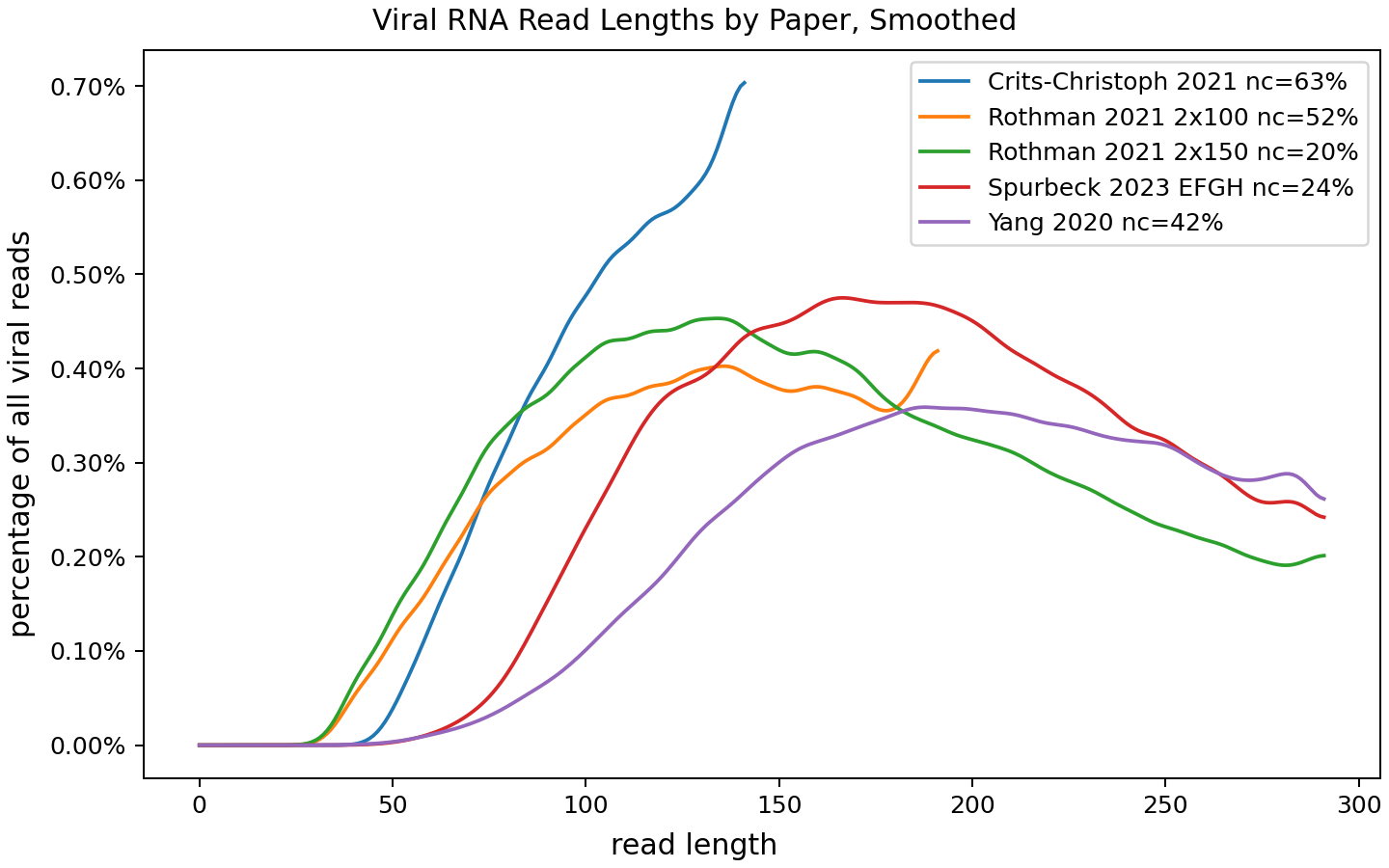
For each paper this is showing the distribution of lengths of reads the pipeline assigned to viruses, in cases where it was able to "collapse" overlapping forward and reverse reads into a single read. [2] The fraction of "non-collapsed" reads (essentially, ones with a gap) is also pretty relevant, and I've included that in the legend. This is why, for example, the area under the purple Yang line can be smaller than the one under the red Spurbeck line: 42% of Yang viral reads aren't represented in the chart, compared to 24% of Spurbeck ones.
Note that for the Spurbeck paper I'm only looking at samples from sites E, F, G, and H: the paper used a range of processing methods and the method used at those four sites seemed to work much better than the others (though still not as well as I'd like).
These charts show how common each length is as a fraction of all viral reads, but what if we additionally scale these by what fraction of reads are viral? If you're studying viruses you probably wouldn't want 50% longer viral reads if in exchange a tenth as many of your reads were viral! Here's the same chart, but as a percentage of all reads instead of just viral reads:
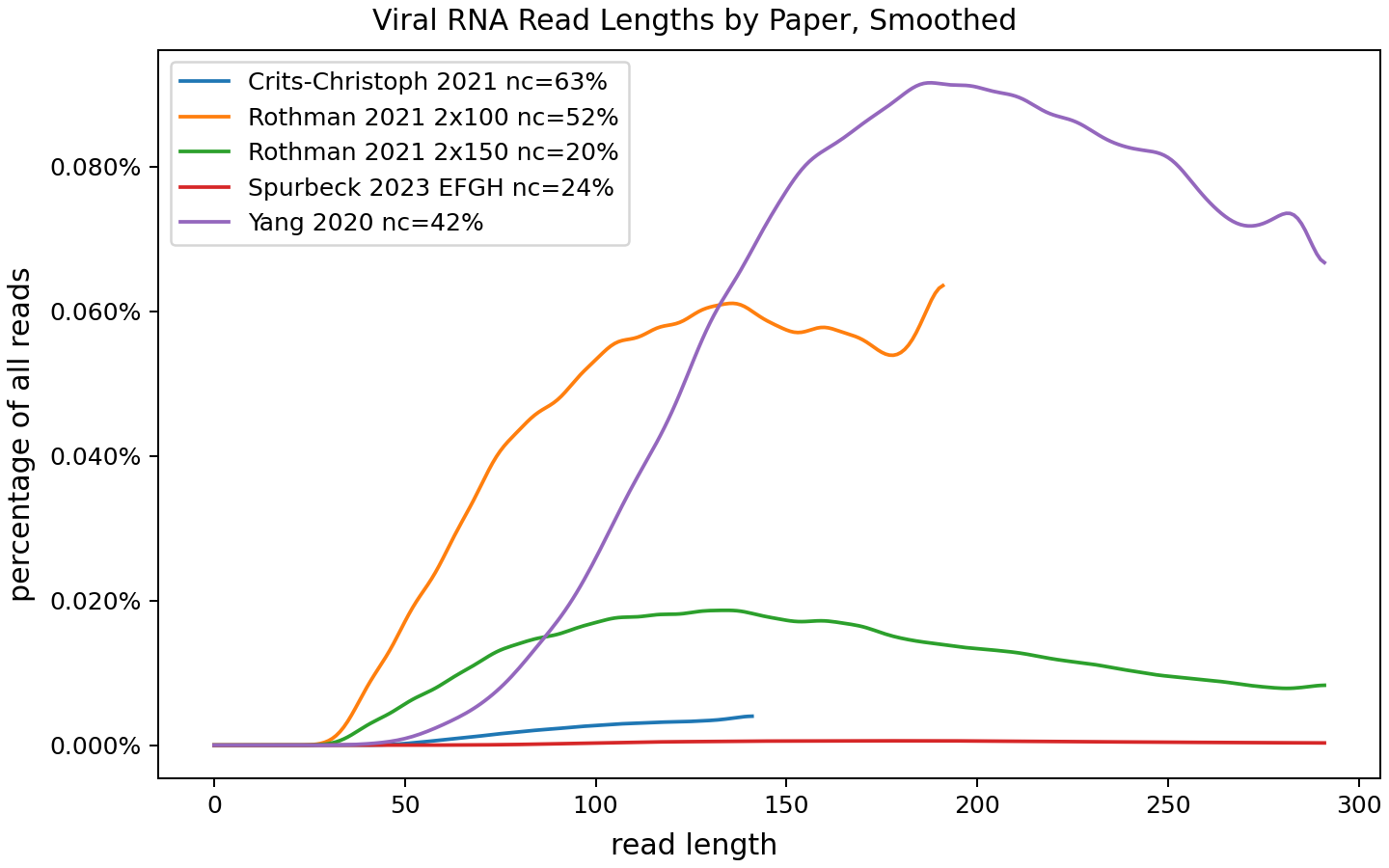
This makes the Yang result more exciting: not only are they getting substantially longer reads than the other papers, but they're able to combine that with a high relative abundance of viruses.
Here's the variability I see between the seven Yang samples:
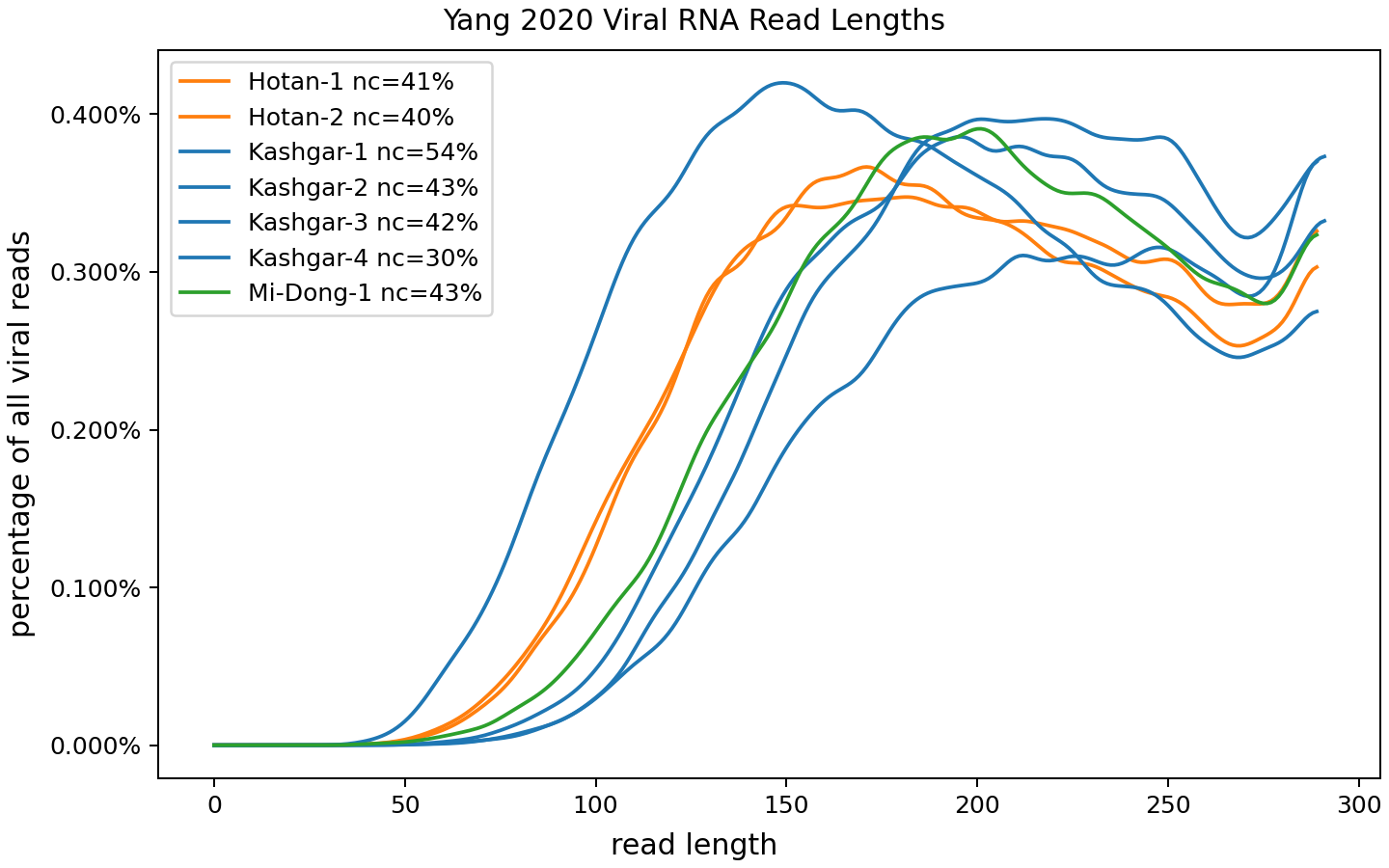
This is also pretty good: some samples are a bit better or worse but nothing that strange.
Now, while this does look good, we do need to remember these papers weren't processing the same influent. There are probably a lot of ways that municipal wastewater in Xinjiang (Yang) differs from, say, Los Angeles (Rothman). Perhaps it's the fraction of viral RNA in the influent, different viruses prevalent in the different regions, the designs of the sewage system, the weather, or any of the many differences between these studies other than their sample processing methods. Still, I expect some does come from sample processing and this makes me mildly optimistic that it's possible to optimize protocols to get longer reads without giving up viral relative abundance.
[1] I don't know what causes this, though I've seen similar length
patterns in a few other data sets including Fierer
et al. 2022, the NAO's sequencing on an Element Aviti, and some
Illumina data shared privately with us.
[2] Ideally I would be plotting the lengths after adapter removal and collapsing only, but because the current pipeline additionally trims low-quality bases in the same step that's not something I can easily get. Still, since Illumina output is generally pretty high quality and so doesn't need much trimming, and what trimming it does need is usually on the "inside", this should only be a small effect.
Comment via: facebook, lesswrong, mastodon, substack