Spending Update 2026
Tags: notyet, money, ea
Every two years (2024, 2022, 2020, 2018, 2016, 2014) I write up our spending. I find it useful to take a step back and really think about where our money is going, and it's not much extra work to get it into a form I can share. I think this kind of information is often useful to people thinking about their own finances, and is undersupplied because of a culture of keeping this kind of thing private. I also like it as a frame for talking through the decisions we've made about what's important to us, since a lot of that is reflected financially. So: where does our money go?Here's our 2025 spending, on a monthly basis:
| Category | pre-tax | post-tax | total |
|---|---|---|---|
| Donations | $0 | $11,874 | $11,874 |
| Taxes | $0 | $2,984 | $2,984 |
| Housing | $0 | $2,022 | $2,022 |
| Childcare | $0 | $3,553 | $3,553 |
| Medical | $226 | $400 | $636 |
| Food | $0 | $964 | $964 |
| Other | $0 | $1,000 | $1,000 |
| Savings | $4,297 | -$6,000 | -$1,703 |
~~break~~ Comparing to previous years and adjusting for inflation:
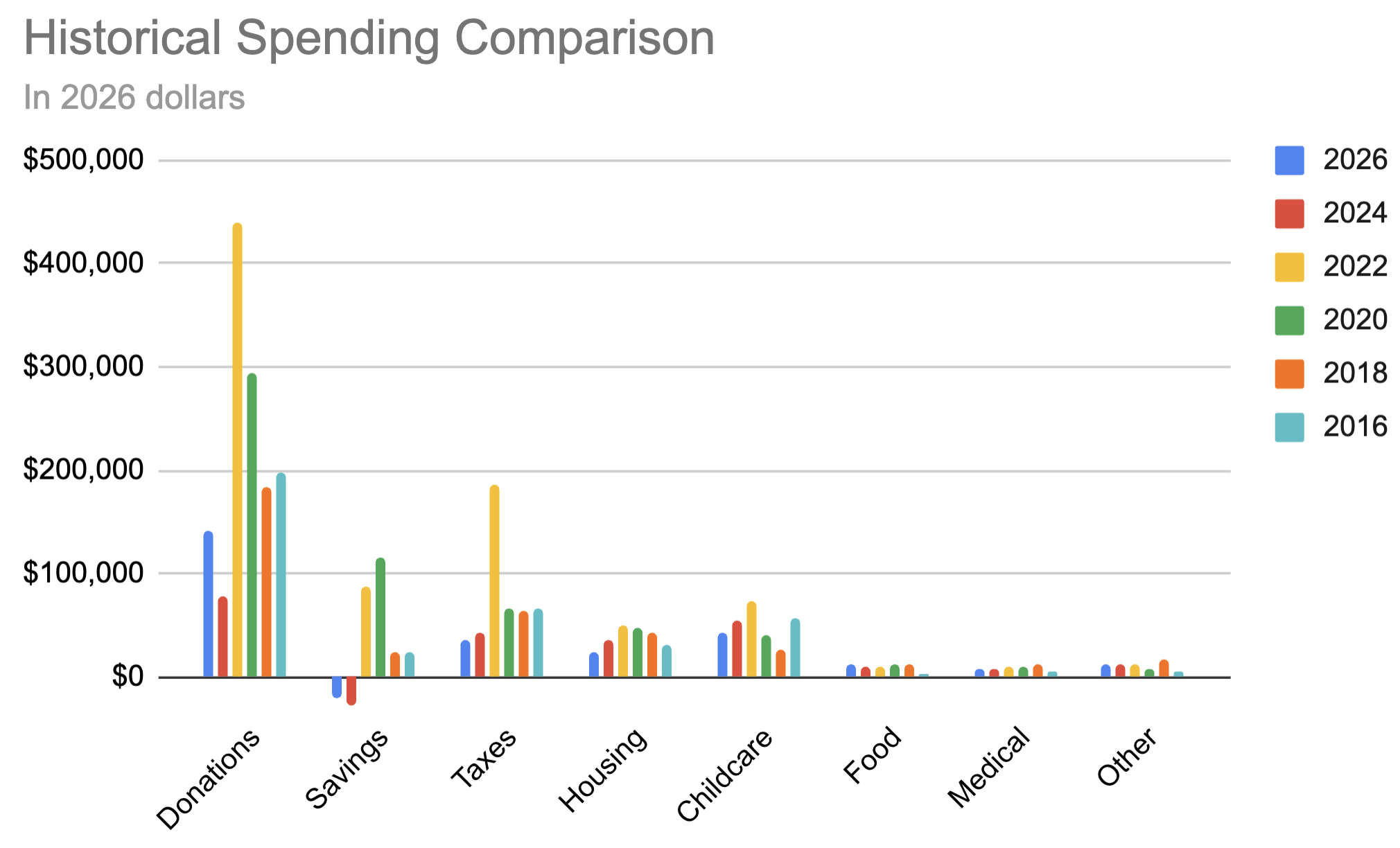
| 2026 | 2024 | 2022 | 2020 | 2018 | 2016 | |
|---|---|---|---|---|---|---|
| Donations | $11,874 | $6,424 | $36,667 | $24,500 | $15,325 | $16,486 |
| Savings | -$1,703 | -$2,344 | $7,222 | $9,625 | $1,948 | $2,027 |
| Taxes | $2,984 | $3,542 | $15,556 | $5,500 | $5,390 | $5,595 |
| Housing | $2,022 | $2,910 | $4,111 | $4,038 | $3,571 | $2,527 |
| Childcare | $3,553 | $4,453 | $6,111 | $3,438 | $2,208 | $4,689 |
| Food | $964 | $763 | $764 | $938 | $974 | $311 |
| Medical | $636 | $671 | $790 | $813 | $969 | $428 |
| Other | $1,000 | $1,042 | $1,111 | $625 | $1,455 | $399 |
Compared to a typical family, the most unusual part is probably how much we're donating. This has been important to us for a long time; for 2025 we decided continue drawing on our savings and increase our giving beyond 50%. Two main reasons, both downstream from the AI boom:
People who've made money in the boom, others will likely be giving more soon. Money spent now can help set up organizations to spend future money more effectively.
This is an especially important window of opportunity. Transformative AI is coming very quickly, for better or worse, and we should push hard for "better".
Looking at it another way, however, the priority we've put on donations has gone down a lot. I'm no longer working at a big tech company, optimizing for high income to maximize donations: we're giving a larger fraction, but of a smaller amount:
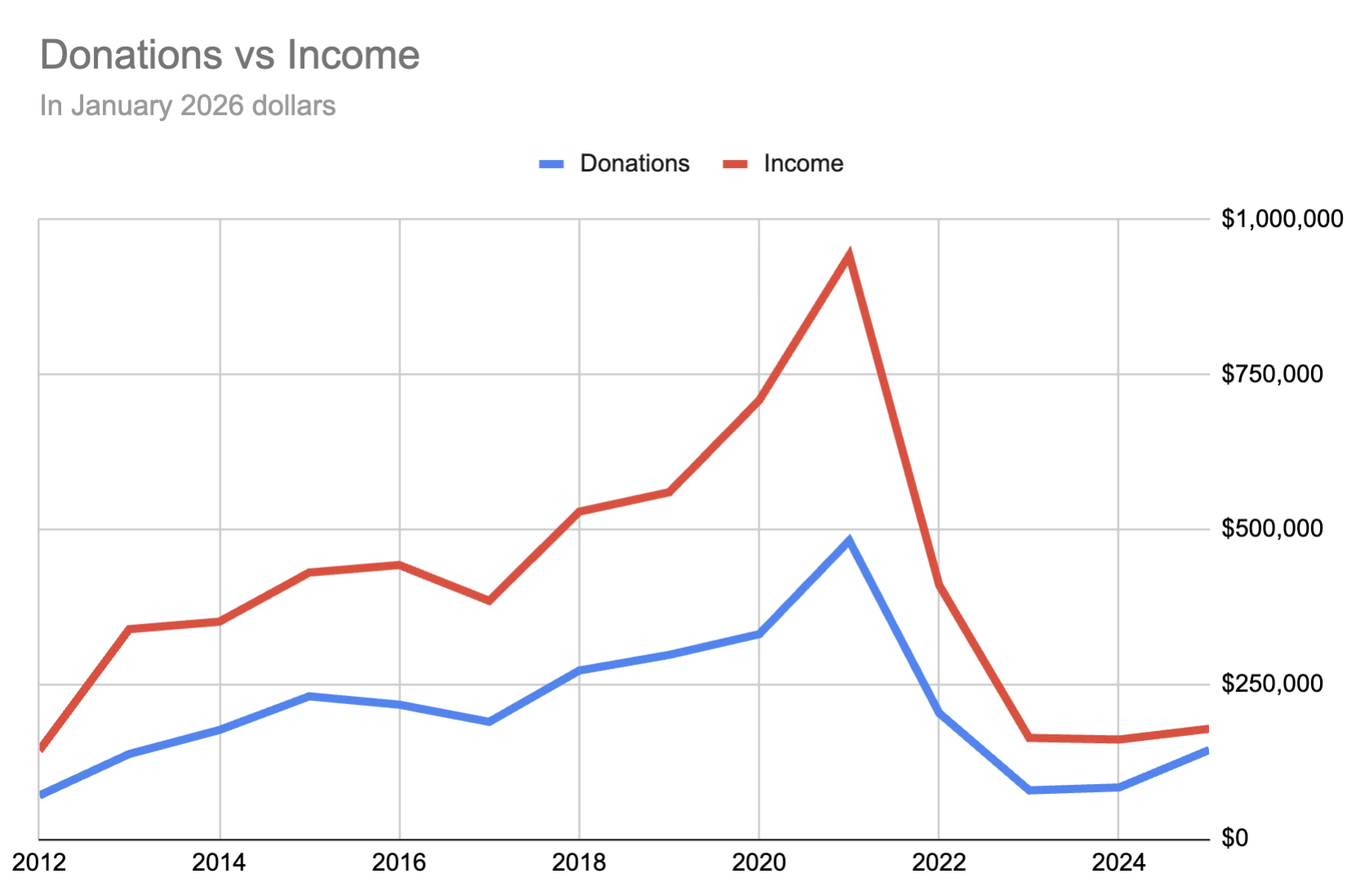
This is a change I feel very good about, since I think my work at SecureBio is really valuable. More than that, while money can enable important work, I see a lot of projects that primarily need a dedicated team to bring them into existence, and I'd be excited for others to move in this direction.
Some things to note:
We're prioritizing directly impactful work. When
We're prioritizing donations.
The biggest changes with 2024 are: Salary sacrifice, then not Pulling donations forwards Medical down higher deductible, less medical visits When I write this post in 2026, what do I expect to be saying?
I think there's a good chance we'll have switched from giving 50% to some form of salary sacrifice. If we do, our pay, donations, and taxes will all be a lot lower.
Childcare should be similar: the nanny share is working and I expect we'll do something similar at least until our youngest starts kindergarten in Fall 2026 (and will show up in the 2028 update).
I'd like to hope I have a better way of accounting for housing and savings in general and have gone back and redone all my previous numbers under the new system, but since that sounds like a ton of work I doubt I'll have done that.
I put about a 10% chance on AI, war, or other major events in this timeframe changing things enough that everything is weird in hard to predict ways.
Donations are up relative to down a lot, because our income is down a lot. We're still going for 50%, though I'm still unsure whether this makes sense given that our workplaces are both altruistically funded.
Savings are down a lot, showing up as negative above. A lot of this is an effect of the amortized approach I'm using for the housing accounting (more). Above I have our housing as $2,800/month, but a lot of that was effectively pre-paid through the downpayment, dormers, mercury spill, solar, heating upgrades, etc. and at the time was counted as 'saving'. While we did spend $14.5k on gut-renovating the first floor bathroom, that's the only really big expense in the last two years: our house expenses have slowed down a lot. And we refinanced in 2021, spreading our remaining payments out over another 30y and dropping our rate from 4% to 3.375%.
All together, if you just look at it on a cashflow basis (how much money left our bank account to deal with housing less how much came back in as rent) over the past two years we've averaged spending of -$442/month. That is, we've brought in enough from rent to cover the portion of our housing that we didn't pre-pay, with a bit left over. This is enough that, on a cashflow basis, we're not spending down our savings and are actually saving a small amount of money.
This all feels awfully messy, but I guess handling this kind of complexity is why accounting is a profession.
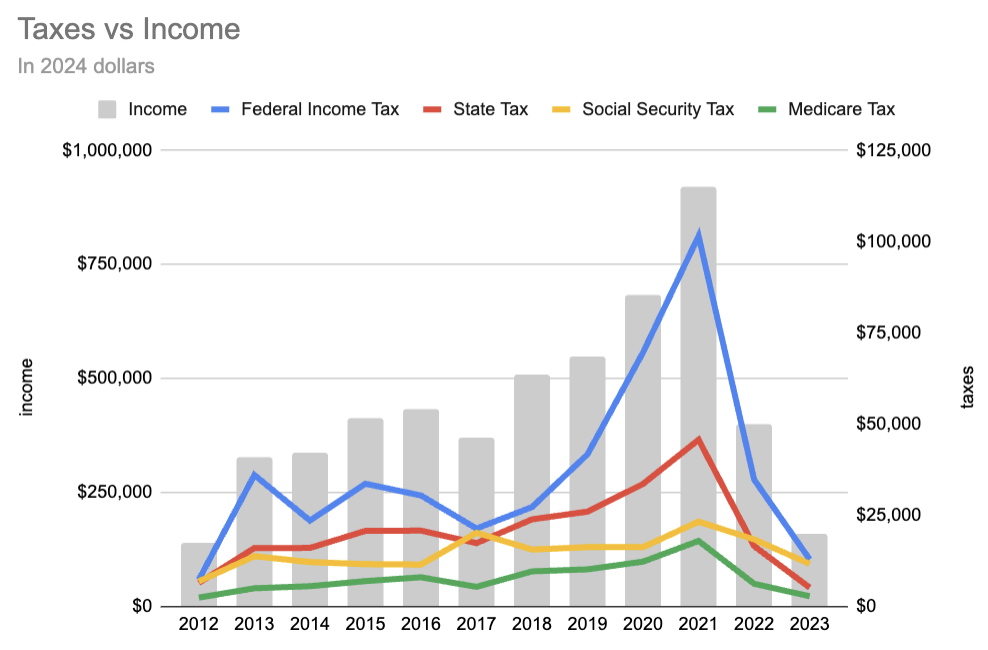
Taxes are much lower. A more detailed view:
Most of what's going on is that we're earning much less money, and so pay less in tax. Our income tax was maybe $5k higher this year than it should have been because half our 2023 donations ended up formally in 2024. It's also interesting that Social Security tax is now a much greater proportion: this is because it's 6.2% of wages up to contribution and benefit base, which was $160k in 2023. Since it's only linear up to that cap, and for many years I was earning more than the cap, it's grown as a fraction of our taxes.
Housing is down a bit because rent has increased (partly Boston getting more expensive, partly renovating and renting out the backyard office).
Childcare is down a bit because we're now doing a nanny share. While the rates we're paying are a bit higher than the end-of-2021 numbers I used last time, each family now pays 2/3 of what they would pay if the nanny were watching only that family's children.
This is the first time I've included inflation in one of these posts, through a combination of it having a larger effect than before and my previously being too lazy to include it.
Details
This post uses the same approach as last year, which is almost the same as before then. Numbers are monthly, based on 2025 spending.